
Since the discovery of graphene, two-dimensional(2D) materials have received considerable attentions due to their outstanding electronic and thermal properties, high specific areas and convenient manufacturing activities. Several graphene-like 2D materials have been fabricated and gained specific applications, such as silicene, phosphorene, MoS2, and etc. In 2011, a new family of 2D transition-metal carbides and nitrides called “MXene” has been fabricated by selective etching of “A” from Mn+1AXn phases (where M is an early transition metal, A is an A-group element, X is C and/or N, and n = 1, 2, or 3). Since Mn+1AXn materials represent a large family that consists of more than 70 members, the corresponding MXenes inherit versatile configurations. To date, more than 15 MXene members have been synthesized by exfoliation of the corresponding Mn+1AXn phases and relatives. The as-synthesized MXenes are typically functionalized by -O, -OH and -F groups. Based on the abundant chemical elements and various functional groups, the physical properties of MXene are significantly dependent on its configuration. Therefore, to better understand and apply these materials, it is crucial to investigate the intrinsic physical properties of the MXenes.
In order to understand the roles of the surface functional groups on the physical properties of MXenes,the group firstly comprehensively studied the structures, mechanical strengths and electronic energy bands of M2CT2 MXenes (M=Sc, Ti, V, Cr, Zr, Nb, Mo, Hf, Ta, W; T=O, F, OH). According to the results, the oxygen functionalized MXenes generally exhibit smaller molar volumes, higher mechanical strengths and more semiconductor members, compared with corresponding MXenes functionalized by hydroxyl and fluorine groups. The stable configurations for most M2CT2 MXenes and the elastic constants c11 are given in Figure 1. The difference of the physical properties in the oxygen, fluorine and hydroxyl terminated MXenes can be ascribed to the higher electron accepting ability of oxygen group. The band gaps of the semiconducting Sc2CT2 (T=F, OH) and M2CO2 (M=Ti, Zr, Hf) MXenes are in the range from 0.85 to 1.8 eV, which are applicable for electronic devices (EPL 2015,111, 26007).
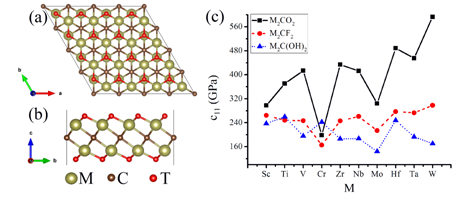
Figure 1 (a) and (b) are the top-view and side-view of M2CT2MXenes, respectively. (c) shows the elastic constants c11 for M2CT2MXenes.
For applying the semiconducting MXenes in electronic devices, the key properties such as carrier mobilities and thermal conductivities of Sc2CT2 (T=F, OH) and M2CO2 (M=Ti, Zr, Hf) are investigated. Sc2CT2 (T=F, OH) present relatively high electron mobilities, especially in its zigzag direction, which value is predicted to be 5.03×103 cm2V-1s-1. Moreover, the electron mobilities are found to be anisotropic, which is due to the special electronic wavefunctions at the conduction band minimum (CBM) as shown in Figure 2(a). Regarding to the thermal conductivity, Sc2CF2 presents a value as high as 472 Wm-1K-1 at room temperature based on a 5 um flake length, and which can be further enhanced by increasing the flake length (Nanoscale 2016, 8, 6110).For the oxygen terminated M2CO2 (M=Ti, Zr, Hf), they all present ultrahigh hole mobilities, which values are of order of 104 cm2V-1s-1. Noteworthily, the predicted high hole mobilities are well consistent with the experimental findings for Ti2COx MXene. The thermal conductivity increases significantly with increasing atomic number of M for M2CO2 (M=Ti, Zr, Hf) MXenes, which can be explained by the enhanced mechanical strengths of MXenes with increasing atomic number of M. The thermal conductivity of Hf2CO2 is comparable with that of pure iron, and that in Ti2CO2 is only 21 Wm-1K-1 (Scientific Reports, 2016, 6, 27971).Based on the predicted data, the semiconducting MXenes could be promising candidate materials for electronic devices.
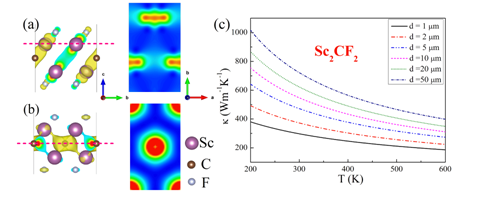
Figure 2 (a) and (b) show the electronic wavefunctions for the conduction band minimum (CBM) and valance band maximum (VBM) respectively for the Sc2CF2MXene. (c) the relationship between the thermal conductivity of Sc2CF2 and the size of flake length in the armchair direction.
Additionally, for the newly synthesized Mo2C MXene, its structural, electrical, thermal and mechanical properties are fully investigated. The MXene is found to present a small molar volume, super-high electrical conductivity, and favorable thermal conductivity. Moreover, it is robust to temperature variations and strains. Explicitly, the electrical conductivity is of order of 106 Ω?1 m?1. The thermal conductivity increases with the increasing temperature; the intrinsic value at room temperature is predicted to be 48.4 Wm?1 K?1, which increases to 64.7 W m?1 K?1 by introducing a heavy n-doping. The thermal expansion coefficient at room temperature is determined to be 2.26×10?6 K?1. The biaxial elastic modulus is predicted as 312±10 GPa by a linear fitting. On the basis of these predicted properties, the Mo2C MXene merits promise for a number of potential applications, such as applying to substrates for other layer materials, and candidate materials for batteries and supercapacitors (Journal of Physical Chemistry C 2016, 120, 15082).
The above work is supported by the National Key Research and Development Program of China (No. 2016YFB0700100), the National Natural Science of Foundations of China (Grant Nos. 11604346,51372046, 51479037, 91226202), the Ningbo Municipal Natural Science Foundation (No. 2016A610272, 2014A610006) and the key technology of nuclear energy, 2014, CAS Interdisciplinary Innovation Team.
Prof. Du Shiyu:dushiyu@nimte.ac.cn
Dr. Zha Xian-Hu:zhaxianhu@nimte.ac.cn
All Images by ![]()